| CHAPTER 1 LEARNING OBJECTIVES | ||
|
|
| ACRONYMS | ||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KEY TERMS | ||
|
Food and Drug Administration (FDA): The FDA is the oldest consumer protection agency regulating a variety of products. Five types of activities can describe the FDA’s regulatory approach: (1) new product review; (2) post-market surveillance; (3) setting standards and writing regulations; (4) scientific research; and (5) regulatory enforcement and corrective action. |
| OVERVIEW OF TODAY'S FDA | ||

|
The Food and Drug Administration (FDA) is a public health agency located within the Department
of Health and Human Services. The FDA is one of the nation’s oldest consumer protection agencies
and has the dual function of law enforcement and scientific research. It has been a law
enforcement agency for over 90 years, and a scientific institution since 1862.
The FDA has approximately 9,000 employees nationwide to monitor the manufacturing, importing,
transporting, storage, and the sale of $1 trillion worth of products (annually). The staff
working in the Washington, D.C. area focuses on new product review and approval,
writing regulatory policy, and safety surveillance of products on the market.
|
| FDA REGULATED PRODUCTS AND RESPONSIBILITIES | ||
|
The products that the FDA regulates are medicinal (drugs, biologics, complex medical devices), radiation-emitting, food (including pet and livestock feed), cosmetics, and the nation's blood supply. The extent of the FDA's responsibility varies based on the product regulated. |
|
|

|
|

|
|
 SAFE WHOLESOME AND SANITARY FOODS - The FDA assesses our food supply to verify that it is free from contaminants. They detect and prevent the spreading of food related infections.
The FDA reviews new food additives for safety before they can be used and sold to the public. They also monitor the safety of dietary supplements, infant formulas, and medical foods.
The FDA is not responsible for regulating meat and poultry products as this falls under the jurisdiction of the United States Department of Agriculture (USDA).
SAFE WHOLESOME AND SANITARY FOODS - The FDA assesses our food supply to verify that it is free from contaminants. They detect and prevent the spreading of food related infections.
The FDA reviews new food additives for safety before they can be used and sold to the public. They also monitor the safety of dietary supplements, infant formulas, and medical foods.
The FDA is not responsible for regulating meat and poultry products as this falls under the jurisdiction of the United States Department of Agriculture (USDA).
|
|
 SAFE COSMETICS - The FDA monitors cosmetics for proper labeling and their safety while in use. However, the FDA does not mandate safety testing of cosmetic ingredients, nor do these ingredients require approval prior to their use.
SAFE COSMETICS - The FDA monitors cosmetics for proper labeling and their safety while in use. However, the FDA does not mandate safety testing of cosmetic ingredients, nor do these ingredients require approval prior to their use.
|
|
 SAFE AND EFFECTIVE ANIMAL DRUGS - The FDA scientifically reviews and evaluates the safety and efficacy data on animal drugs used by Veterinarians and either approves or disapproves their use.
The FDA also evaluates the safety of drugs used with livestock and their effects on the environment and the consuming publics' health.
SAFE AND EFFECTIVE ANIMAL DRUGS - The FDA scientifically reviews and evaluates the safety and efficacy data on animal drugs used by Veterinarians and either approves or disapproves their use.
The FDA also evaluates the safety of drugs used with livestock and their effects on the environment and the consuming publics' health.
|
|

|
|

| FDA'S REGULATORY APPROACH | ||
|
The FDA’s regulatory approach varies based on the respective product. There are five types of activities that can describe its approach. The activities consist of new product review, surveillance, setting standards and writing regulations, performing scientific research, regulatory enforcement, and mandating corrective action. |
|
NEW PRODUCT REVIEW - The core of the FDA’s public health duties is reviewing new products such as drugs, biologics, complex medical devices, and food additives. The FDA's regulatory approach for reviewing new medicinal products can be divided into two types of activities: its pre-approval review and post-approval surveillance. The pre-approval activities involve performing a scientific review and evaluation of the safety and efficacy data submitted in a manufacturer's medicinal product marketing application. The FDA evaluates the study results of laboratory, animal, and human testing to determine whether the resulting benefits outweigh the risks associated with the use of the product. The post-approval activities involve the use of the FDA's post-marketing surveillance system to assess the approved medicinal product's ongoing safety while it is in use (Refer to Surveillance Description). In addition, the FDA performs a safety review and grants approval for new food and color additives, infant formulas, medical foods, and animal drugs. |
|
SURVEILLANCE - The FDA performs two types of surveillance activities on products approved for marketing. One is through its post-marketing surveillance reporting system and the other is through its on-site facility inspections. The post-marketing surveillance reporting system consists of the manufacturers mandatory reporting requirements and the voluntary reporting by consumers and health care providers of adverse experiences and product problems. Every year the FDA receives approximately 400,000 of these reports for its review and management. The FDA performs on-site inspections of manufacturers to verify that the products produced have met the appropriate quality control manufacturing standards.
|
|
STANDARDS AND REGULATIONS - The FDA establishes regulations and product quality control standards to set “benchmarks” in defining the requirements for manufacturers to conduct valid and ethical scientific research, manufacture quality products, adequately label products, and the mandatory reporting of a products ongoing safety while in use. Before product approval the regulations set scientific standards in the conduct, implementation, monitoring, and evaluation of research in humans. It establishes ethical standards and safeguards for research involving human participants. After product approval it sets standards in the mandatory reporting requirements of adverse events and product problems to the FDA. In addition, the FDA assists foreign governments in producing and importing products that have met standards equivalent to those enforced in the United States. |
|
RESEARCH - The FDA performs scientific research to provide the basis for its regulatory decisions and to establish the methods necessary to assess risks. The research results guide the FDA in setting standards and writing regulations. |
|
ENFORCEMENT AND CORRECTIVE ACTION - As part of the FDA's law enforcement function, it performs on-site inspections of the manufacturer's facilities to assess their regulatory adherence. The FDA's action against manufacturers in violation of the regulations depends on the severity of the violation. One action involves working with the manufacturer to correct the problem voluntarily. If voluntary corrections do not occur, or the violation is more severe, the FDA can impose legal remedies, such as product recall, product seizure by federal marshals, detaining of imports, or criminal penalties. |
| FDA DESIGNATION AND LOCATION TIMELINE | ||
|
Figure 1.1 illustrates the milestones of the Food and Drug Administration (FDA) establishment and designation as an agency in the United States Federal Government. |
|
|
FDA Designation and Location TimeLine - Figure 1.1 |
| THE KEY FDA CENTERS THAT REVIEW AND APPROVE MEDICINAL PRODUCTS | ||
|
The FDA resides under the Department of Health and Human Services. Within the FDA there are several centers with specialized functions for a particular area. Figure 1.2 illustrates the centers responsible for the review and approval of medicinal products. |
|
|
Department of Health and Human Services Medicinal Product Centers - Figure 1.2 |
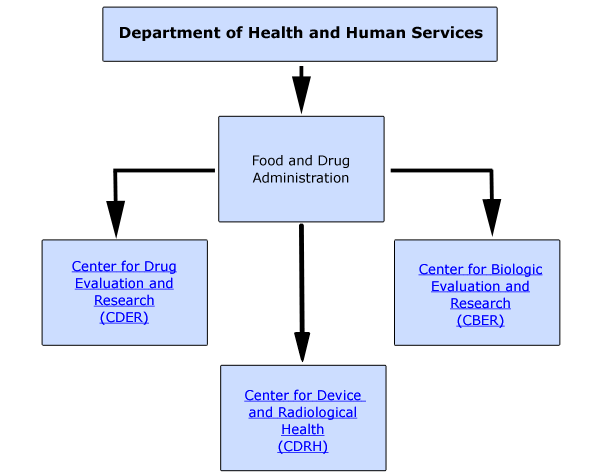
|
|||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||

|
TABLE OF CONTENTS |